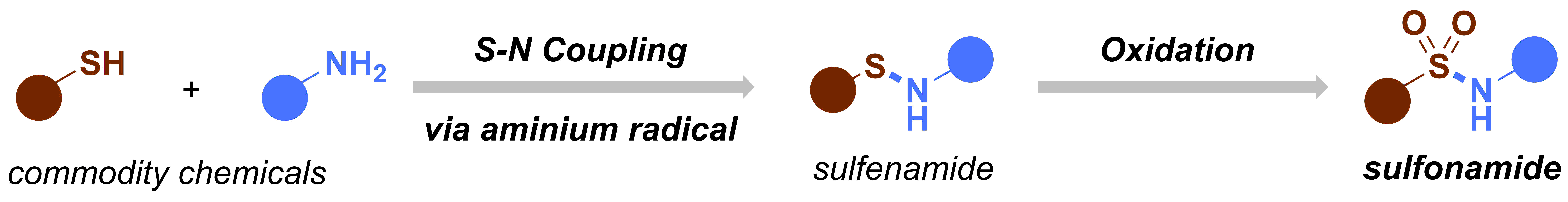
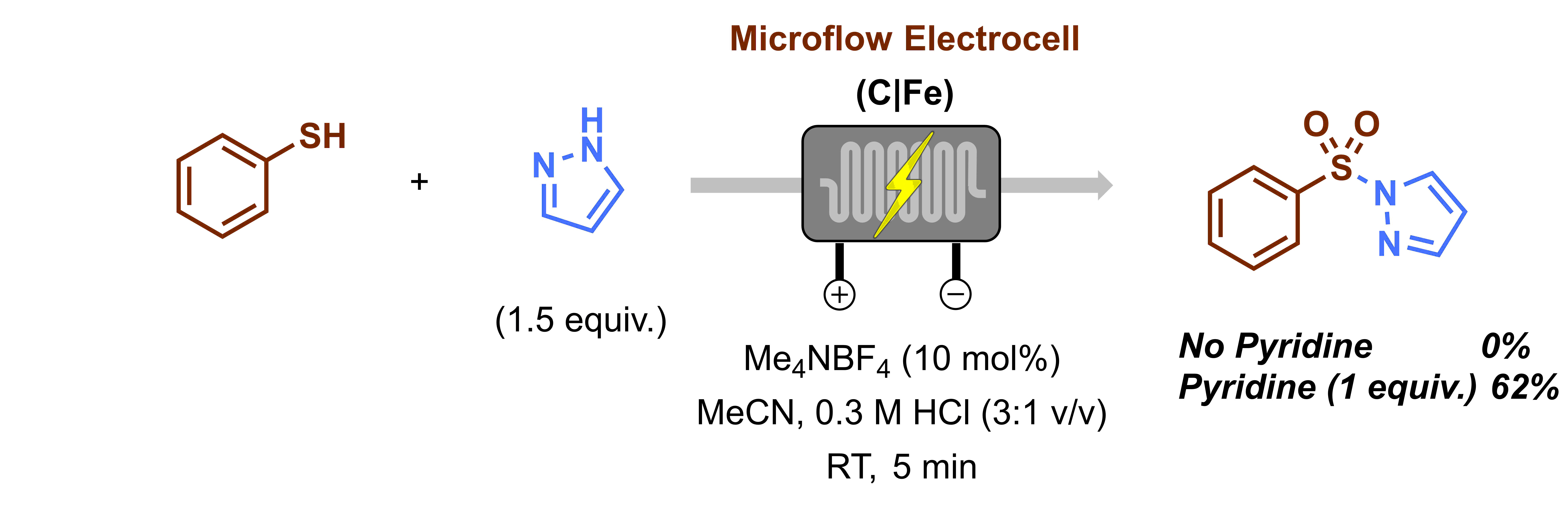
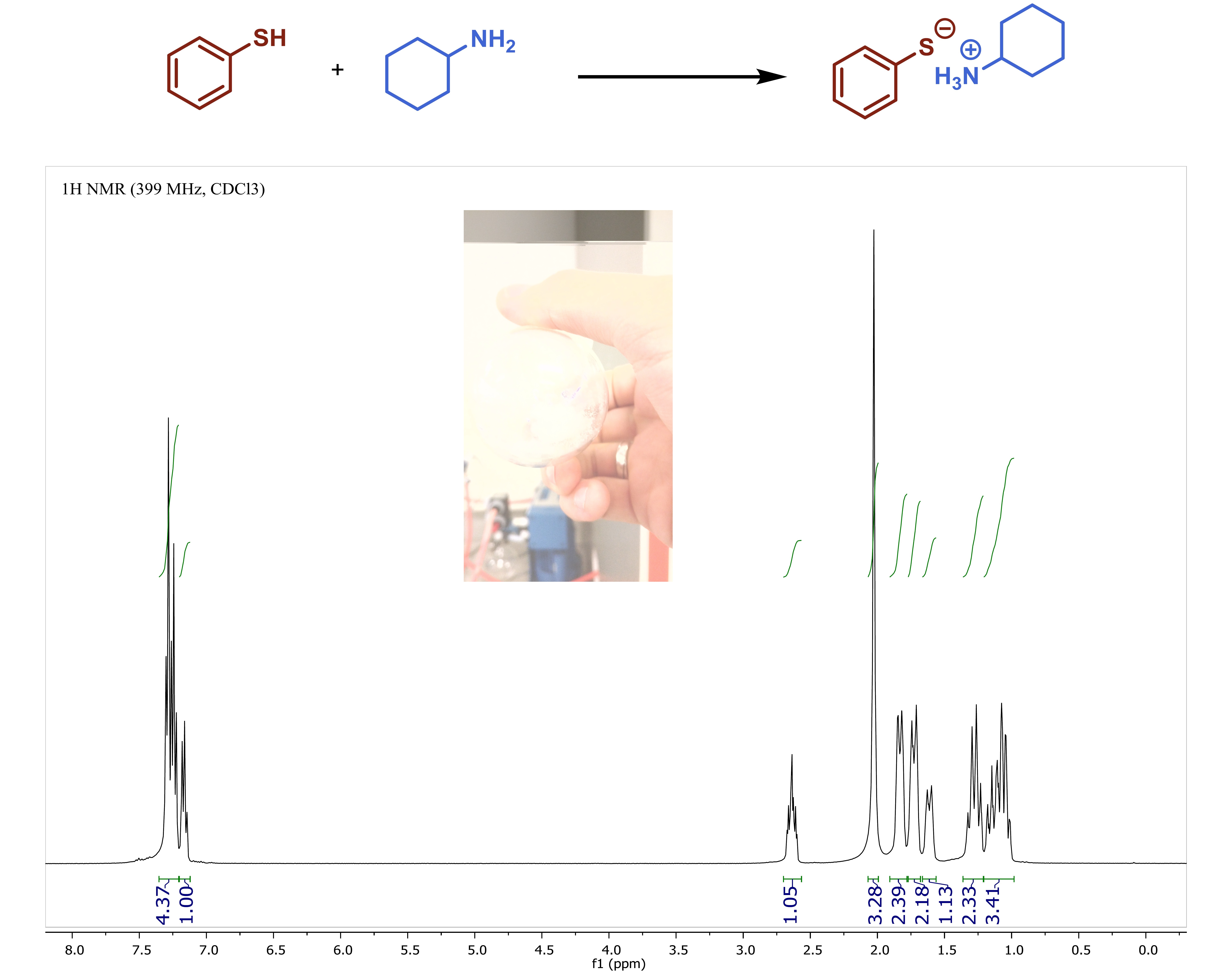
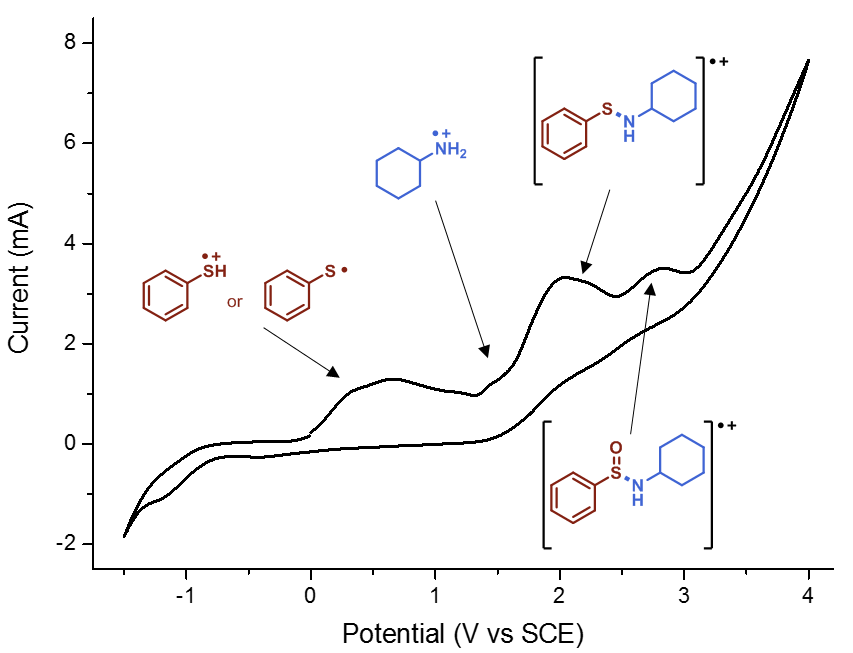
When I embarked on my independent academic career, my mission was clear: to push the boundaries of synthetic organic chemistry through technological innovation. One of my most audacious dreams was to create a chemical robot capable of working alongside human chemists, alleviating the burden of arduous and time-consuming tasks, such as reaction optimization. However, from the outset, I knew that this endeavor would be anything but easy. The examples I found in the literature appeared prohibitively expensive, requiring a diverse team with a wide range of skills, and the assembly process would be a time-consuming challenge in itself. Could we realistically pull this off? At the dawn of our research group, the answer was a resounding ‘no.’ Our team was small (1-5 PhD students in the first five years), resources were limited, and the risks loomed large: the potential for burning through funds and human capital without any publication to show for it. At that time, this was a risk we simply couldn’t afford to take.
The assembly of the team
Confidence in embarking on robotic projects began to take shape gradually after we successfully published an automated strategy for conducting Stern-Volmer experiments.[1] This platform, while relatively simple and cost-effective, made a significant scientific impact by delivering highly reproducible results for these notoriously finicky kinetic experiments.
Enter Zhenghui Wen: When I welcomed in September 2018 Zhenghui Wen, a skilled chemical engineer from China, to our team, I explained our ambitious vision. Zhenghui’s immediate enthusiasm ignited a flurry of activity. He began 3D-printing a multitude of reactors and liquid handlers while also diving into programming Arduino drivers. However, it soon became evident that the RoboChem project couldn’t be a one-person endeavor — though Zhenghui did exceptional work. We made the challenging decision to temporarily halt the project.
Around 2020, the tides began to turn in our favor. Our success in securing funding improved substantially, and our research group underwent remarkable growth. Additionally, I received a valuable startup package at the University of Amsterdam, enabling the purchase of a benchtop NMR, a pivotal yet costly instrument that would provide critical data on yield and selectivity.
Aidan Slattery Joins the Team: Around September 2020, Aidan Slattery, an Irish chemist with invaluable flow experience gained at SnapDragon, became a vital addition to our team. His dedication was fully committed to realizing the RoboChem vision.
Pauline Tenblad’s Expertise: Just a few months later, we were fortunate to welcome our first Swedish PhD student Pauline Tenblad. Her expertise in chemical engineering was crucial for the machine learning efforts required to enable self-optimization within the RoboChem platform.
Dr. Diego Pintossi Takes the Helm: Joining the team alongside Pauline was Dr. Diego Pintossi, a versatile, Italian chemical engineer. His role involved mentoring, coordinating efforts, and ensuring a seamless and productive journey toward our objectives.

Picture 1. Meet the Pioneers: The Original RoboChem Team in Front of the Platform (left to right): Aidan Slattery, Zhenghui Wen, Pauline Tenblad, and Diego Pintossi.
Assembling Hardware and Seamlessly Integrating it with Code: Bringing the RoboChem Vision to Life
These four exceptional researchers embarked on the formidable task of assembling the intricate hardware components and executing the essential coding for our project. The challenges we encountered during this phase were nothing short of daunting.
One major challenge we grappled with was the need for precise control over every piece of equipment through software integration (Figure 1). This necessitated the development of a sophisticated control system to ensure precise functionality of each component. Hardware-related hurdles further compounded our efforts, with compatibility issues and the need for meticulous fine-tuning of mechanical components becoming recurring challenges. For instance, we faced ‘lost in translation’ moments between our 64-bit software control and 32-bit hardware. Switching between these applications took Pauline a few days to untangle. There were countless other small, not-so-glorious issues to resolve, many of which won’t even make it into the final paper.
As progress proved slow and tangible results were limited, the paramount task at hand became maintaining focus and motivation. Tim den Hartog, a visiting researcher from Zuyd University of Applied Sciences, and I took it upon ourselves to keep the team’s spirits high throughout this challenging phase. Together, we actively participated in the biweekly ‘RoboChem meetings,’ where we discussed our ongoing progress and offered suggestions to overcome some of the hurdles that we encountered.
The combined effort of coding and hardware installation stretched over an arduous 2-3 year period, emphasizing the monumental nature of our undertaking. You might wonder why we didn’t opt for readily available off-the-shelf units. The answer, though deceptively simple, lies in the flexibility required for our highly specific applications. Most commercial units function as black boxes, lacking the adaptability needed for our unique project demands. Tailoring such units to our requirements would have presented nearly insurmountable challenges.

Figure 1. Embarking on the First Automated Steps: Taking the initial strides in automating syringe pumps and mass flow controllers, and implementing Arduino controllers.
The First Test: From Stress to Relief, A Remarkably Smooth Journey
But once all the bugs were meticulously ironed out of our system, we stood on the precipice of a crucial milestone—the first test (Figure 2). Our focus honed in on photocatalysis, a domain in which our group has ample experience. We decided to implement one of our versatile photochemical flow reactors within RoboChem, complete with a set of high-intensity LEDs.[2] This allows us to tune the light intensity meticulously, a feature we think might be important for the optimization of photocatalytic transformations.

Figure 2. The RoboChem Platform: Fully Assembled and Primed for Inaugural Testing.
I must admit that during this critical phase, I took a step back from the lab, preferring not to add any additional stress to my dedicated team. However, behind that cautious facade, I was filled with eager anticipation. You see, the stakes were exceptionally high. If this inaugural test failed to yield results, it would have meant a significant investment of time wasted, not to mention the daunting task of restoring the team’s confidence. [The specter of potential failure loomed heavily over us.]
On July 21, 2022, as I went about my day, a notification chimed on my phone—a WhatsApp message from Aidan. It was a moment that would define our journey (See Figure 3). The message held the key to our hopes and fears. The results were nothing short of astonishing: the experiment worked seamlessly, and we achieved an unprecedented level of optimization. We had successfully executed a photocatalytic hydrogen atom transfer-induced Giese-type radical addition to benzalmalonitrile. The euphoria that swept through our team was palpable, and in that instant, we knew we were on the cusp of something remarkable.
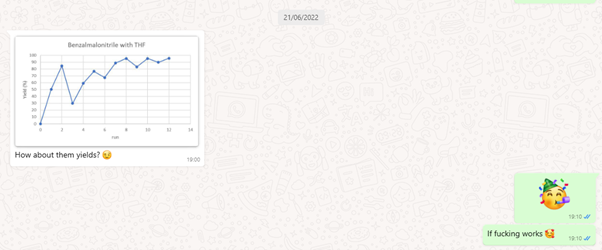
Figure 3. WhatsApp message from Aidan, filled with excitement, prompts an exuberant response from Tim, signaling a groundbreaking moment in the RoboChem project [pardon the F-bomb from Tim].
Progressing Forward: Exploring Diverse Case Studies
Once we obtained those initial results, and I assure you, this was truly a first result (no joke), we realized that we held something incredibly valuable in our hands. We also understood the importance of showcasing the full potential of the RoboChem platform. If we could achieve that, we knew we might be holding the key to a top-tier paper. I instructed the RoboChem team to explore the system thoroughly, with the understanding that once we comprehended its capabilities, we would be able to devise a strategic plan for our next steps.
Around November 2022, I reached out to Zhenghui and Aidan, suggesting that we select a diverse array of photocatalytic synthetic methods and initiate a small-scope optimization, typically with around five examples per method. It was at this stage that our Spanish postdoc Jesus Orduna, who was already a year in our NRG team, joined the RoboChem team, bringing his expertise to assist in chemistry selection, results analysis, and compound isolation.
Our working hypothesis was that while chemists often develop a single set of conditions and apply them universally across the substrate scope, individual AI refinements tailored to each molecule could yield superior results. Additionally, we were well aware of the challenges associated with photocatalysis. This activation mode is notoriously complex, involving intricate photophysics and poorly understood mechanisms. Moreover, technological issues, such as setup variability and irradiation profiles, contribute to interlaboratory irreproducibility, making it a time-consuming endeavor to achieve consistent and reliable results. In other words, the ideal testcase to validate RoboChem!
To our delight, we found that across all examples, spanning hydrogen atom transfer photocatalysis and photoredox catalysis, the RoboChem platform either matched or significantly surpassed yields reported in the literature. It often surprised us by making nuanced choices, such as opting for very low light intensity when beneficial for a specific molecule, while favoring higher intensities in other cases. The data yields more intriguing conclusions, all of which are elaborated in the paper. Notably, for each substrate, we compiled comprehensive data sets with results spanning from low to high yields, providing a clear picture of the parametric sensitivity for each molecule. This approach, we believe, sets our research apart as in literature typically only the best results are highlighted.
The importance of reproducibility
While the results exceeded our wildest dreams, it was imperative to eliminate any room for error. To achieve this, we implemented a rigorous quality assurance process. We utilized the best reaction conditions and ran them consistently in the same photochemical flow reactor, yielding up to 5 mmol of product in each case. Aidan, Zhenghui, and Jesus took on the demanding task of isolating all the compounds, with the obtained yields closely mirroring those achieved with the inline benchtop NMR (Picture 2). Subsequently, each compound underwent comprehensive characterization through NMR, MS, and other analytical techniques. Although time-consuming (we believe that this human isolation and characterization phase served as the rate-determining step, as the entire optimization protocol was fully automated and hands-off), it provided us with unwavering confidence that the results delivered by RoboChem were genuine and not a mere fluke.

Picture 2. Capturing Dedication: Jesus and Aidan, sleeves rolled up, diligently isolating the final compounds.
Paper Submission: The Waiting Game Begins…
Once we had gathered all the data, the exciting journey of crafting our paper began. The initial draft took shape in the hands of Aidan and Pauline, while Zhenghui worked his magic to create the graphics that would highlight our findings. When they presented me with their rough draft, I couldn’t help but feel a surge of enthusiasm. This phase, where you take the collective hard work and start shaping the narrative, is one of the most enjoyable aspects for me.
Typically, I maintain very close contact with the team during this process, bombarding them with multiple questions every day, whether in person or via WhatsApp. We delve into intricate details and fine-tune the storyline. This collaborative refinement process, however, demands time and dedication. For a paper as multifaceted as this one, integrating various disciplines such as chemistry, chemical engineering, programming, machine learning and robotics, it takes me at least two weeks to craft a narrative that we can all take immense pride in.
During the revision process, the team dedicated themselves to creating a comprehensive Supporting Information section, meticulously detailing procedures, characterizations, and optimization tables.
Once everyone was satisfied with the manuscript, we submitted it to ChemRXiv[3] and I began presenting it at conferences. The first opportunity arose at the 6th Flow Chemistry and Continuous Processing Conference in Boston (May 2-3, 2023). The prospect of sharing our work in the city where my journey began, learning flow chemistry at the Buchwald Research Group at MIT, just a 5-minute walk away, filled me with excitement. After my presentation, I was inundated with questions, particularly about RoboChem. The enthusiasm from the audience remained consistent throughout subsequent talks. I even made a playful remark to the RoboChem team that, while I was joking, held some truth: if I wanted questions on other parts of my talk, I’d have to remove RoboChem from the slides.
This overwhelmingly positive feedback boosted our confidence to aim high and submit the paper to the most prestigious journals in our field. We set our sights on Science (See Figure 4A). The submission was met with anticipation and nervous excitement. A promising sign emerged when the paper was sent out for review, spotted in the portal by Aidan (See Figure 4B). Then, on July 31, an email from the Science editor arrived, containing the feedback from the referees. The feedback was predominantly positive, and I couldn’t contain my excitement when I shared the news with the RoboChem team (See Figure 4C). Two referees had provided feedback with an acceptance recommendation, while one referee had a few specific requests, the most notable being, ‘Can you explore dual catalytic pathways, as these are notoriously challenging to optimize?’
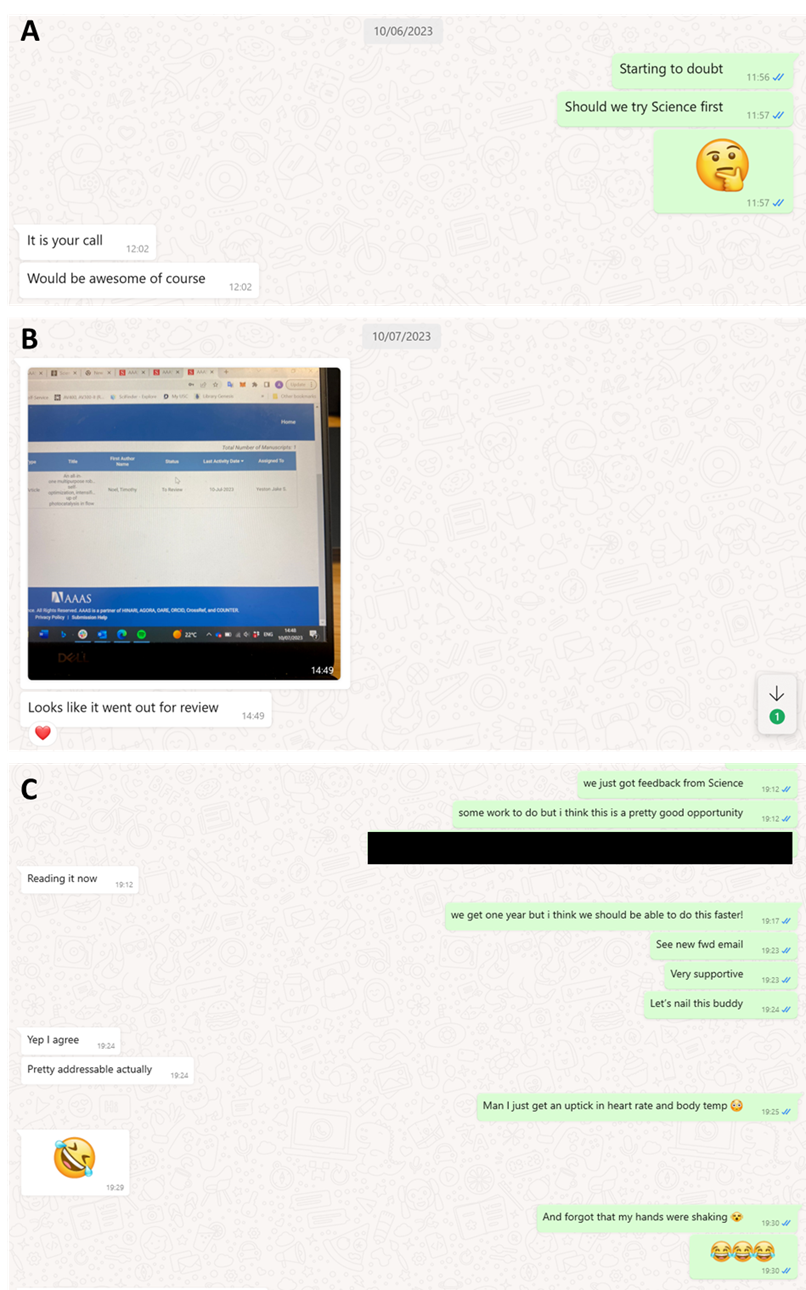
Figure 4. Snapshot of key WhatsApp conversations between Tim (in green) and Aidan (white): (A) Should we try to submit to Science? [June 10, 2023] (B) Paper sent out for review, excitement gets built up. [July 7, 2023] (C) Feedback is in, two yes and one conditional feedback. We are in business, however, more work to do! [July 31, 2023]
The Final Stretch: Let’s Ace This One…
When you receive such exceptional feedback, two emotions intertwine: excitement and nerves. On one hand, the excitement of receiving fantastic feedback and the prospect of a potential paper in Science ignite your spirits. On the other hand, the nerves creep in; you realize you can’t afford to squander this remarkable opportunity. The twist was that we were all on vacation when this happened, so we couldn’t dive into action right away. Aidan and Jesus had to handle things in the lab, and Zhenghui, who had been an integral part of our team, had returned to China, become a father and was applying for jobs. In fact, he even pondered, ‘Should I come back? I am willing to do this!’ This underscores the unwavering dedication of our team to see this through.
Once our vacation ended, and we had some time during the break to strategize, we swiftly concurred that we needed to perform the experiments requested by referee 3. We opted for a photocatalytic cross-electrophile coupling, a realm we had recently explored and understood well.[4] This familiarity instilled confidence that we could execute it successfully. However, our initial attempt didn’t yield promising results; our NMR showed virtually nothing. It was at this point that Jesus’s background in organometallic chemistry came to our rescue. He suspected the interference of paramagnetic nickel, which was muddling the NMR signals. Indeed, by introducing a slight excess of ligand, we managed to prevent the formation of this nickel species and finally observed the NMR signals essential for quantification.
At this stage, we decided to go all out, exploring an extensive chemical space, incorporating both continuous and discrete variables. To our astonishment, the results were nothing short of spectacular, surpassing our original conditions by substantial yield margins and offering a plethora of unique insights (thank you referee 3! Your suggestion proved one of the most compelling cases we carried out on RoboChem). We once again meticulously isolated the compounds to validate the RoboChem results and submitted a revised version along with a rebuttal addressing all the questions. And then, the waiting game commenced.
While our confidence was high upon submission (we had diligently fulfilled all their requests), with each passing week, our anxiety grew (Had we truly covered everything? Why was it taking so long?). Aidan and Zhenghui kept refreshing the portal incessantly; minor updates appeared, but no email landed in my inbox. Sleep became elusive, as I frequently woke up (considering the 6-hour time difference with the USA, could the email arrive during our night?). Finally, on November 20th, while I was aboard the TGV to Lyon, I opened the long-awaited email from the editor. The news was favorable; referee 3 acknowledged that the new results were indeed compelling, albeit referee 2 had some additional questions. Fortunately, these seemed to revolve around textual changes, sparing us from further experimental work. We invested ample time to address them to the best of our ability and submitted the revised version within two weeks. On December 13th, the ultimate acceptance letter from the editor arrived, and it was an immensely gratifying moment (see Figure 5). While we had anticipated the acceptance by then, receiving the final verdict was still a heartening relief.
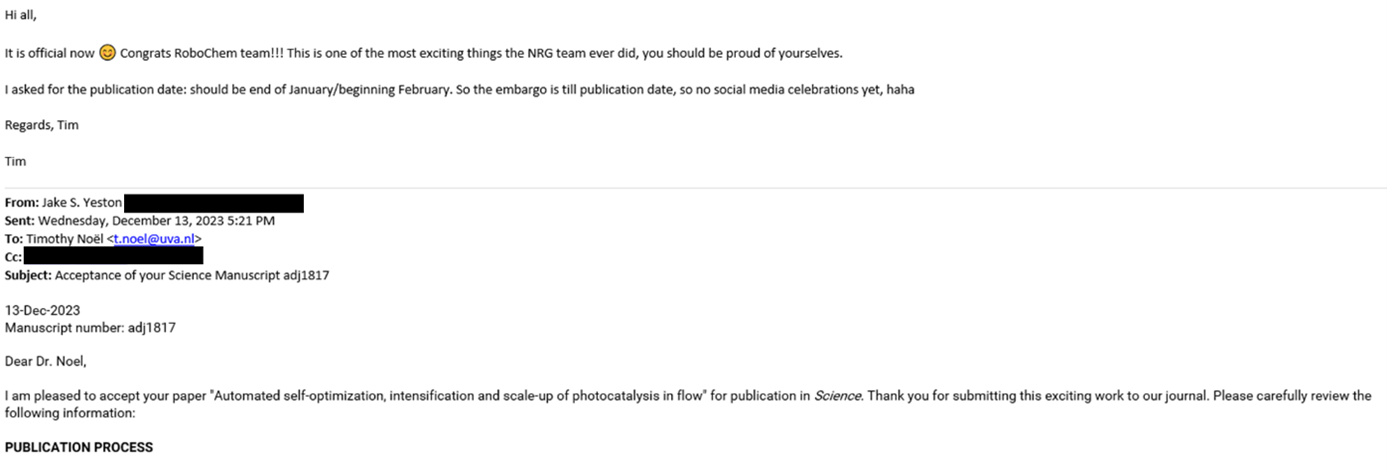
Figure 5. The Elation of the Final Acceptance Letter and the Joyous Forward to the RoboChem Team.
Some last remarks…
As we stand here today, a few weeks after the fact, our paper has been published online by Science.[5] The sense of accomplishment still lingers, and our team is tirelessly working on exploiting and expanding the RoboChem platform for new applications. We sincerely hope that you find our paper and the story behind its creation as enjoyable as we do.
While the conclusion is a happy one, the journey to reach this point was marked by numerous ups and downs. We encountered setbacks, moments of stress, and challenges that tested our resolve. By offering you this behind-the-scenes account, we aim to provide you with a genuine glimpse into the genesis of this work and our approach to projects within our research group. It’s crucial to recognize that this success was the result of dedicated individuals, each possessing unique and complementary skills. We embarked on this journey without full foresight of how it would unfold, and it was during the assembly of various components that the platform revealed its true potential.
Tim Noël
Amsterdam, 25/01/2024
[1] Kuijpers K.P.L.; Bottecchia, C.; Cambié, D.; Drummen, K.; Koenig, N. and Noël, T. A fully automated continuous‐flow platform for fluorescence quenching studies and Stern‐Volmer analysis. Angewandte Chemie International Edition 2018, 57(35), 11278-11282 DOI: 10.1002/anie.201805632.
[2] Wan, T.; Wen. Z.; Laudadio, G.; Capaldo, L.; Lammers, R.; Rincón, J. A.; García-Losada, P.; Mateos, C.; Frederick, M. O.; Boroersma, R. and Noël, T. Accelerated and Scalable C(sp3)–H Amination via Decatungstate Photocatalysis Using a Flow Photoreactor Equipped with High-Intensity LEDs. ACS Central Science 2022, 8 (1), 51-56 DOI: 10.1021/acscentsci.1c01109
[3] Slattery, A; Wen, Z; Tenblad, P.; Pintossi, D.; Sanjose-Orduna, J.; den Hartog, T. and Noël, T. An all-in-one multipurpose robotic platform for the self-optimization, intensification and scale-up of photocatalysis in flow. ChemRxiv, 2023, DOI: 10.26434/chemrxiv-2023-r0drq
[4] Luridiana, A.; Mazzarella, D.; Capaldo, L.; Rincón, J. A.; García-Losada, P.; Mateos, C.; Frederick, M. O.; Nuño, M.; Buma, W. J.and Noël, T. The Merger of Benzophenone HAT Photocatalysis and Silyl Radical-Induced XAT Enables Both Nickel-Catalyzed Cross-Electrophile Coupling and 1,2-Dicarbofunctionalization of Olefins. ACS Catalysis 2022, 12 (18), 11216–11225 DOI: 10.1021/acscatal.2c03805
[5] Slattery, A; Wen, Z; Tenblad, P.; Sanjose-Orduna, J.; Pintossi, D.; den Hartog, T. and Noël, T. Automated self-optimization, intensification and scale-up of photocatalysis in flow. Science 2024, DOI: 10.1126/science.adj1817
 Figure 1. Email received from the European Commission.
Figure 1. Email received from the European Commission. Figure 2. Screenshot from the Evaluation Letter
Figure 2. Screenshot from the Evaluation Letter Figure 3. The so-called president letter with the clear message: Proposal funded.
Figure 3. The so-called president letter with the clear message: Proposal funded.



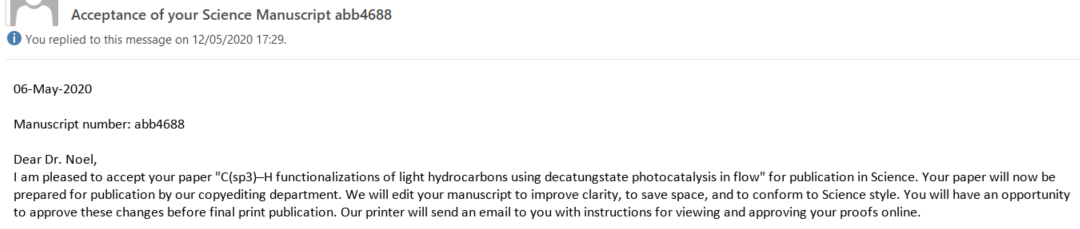
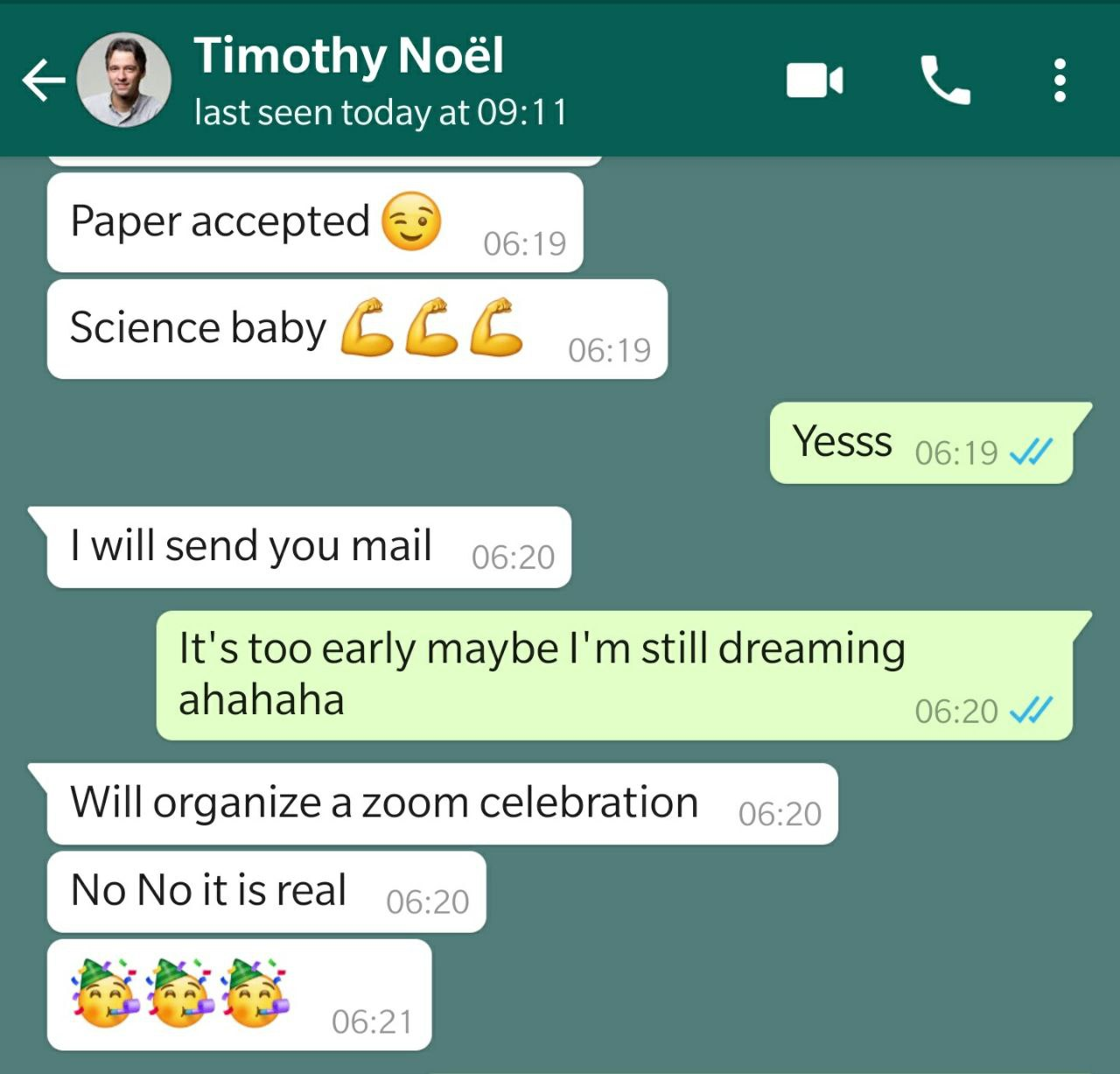
 Figure 1
Figure 1