
With great joy we’d like to share the news that our latest work on the Visible Light-Mediated Arylation of Cysteine was just published yesterday in Angewandte Chemie International Edition (DOI: 10.1002/anie.201706700). Recently, it became somewhat of a tradition in our group to write a short blog on the behind-the-scenes of our papers. So here’s all about the “visible light-mediated arylation of cysteine” ☺.
The beginning
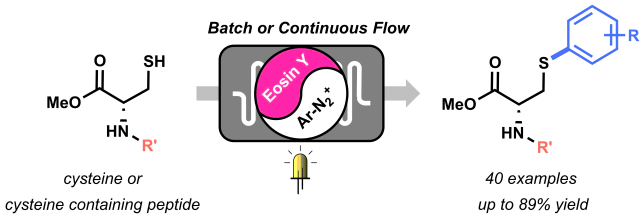
The beginning of this project coincided with the start of my PhD in the Noёl research group, back in 2015. Right from the start, Tim and I agreed we wanted to apply the expertise of his group on photoredox catalysis to new methodologies for biomolecule modifications. But how to do it? Following on the recent findings from Tim and my former colleague Natan Straathof, we knew thiols are efficient moieties in trapping radicals generated via photoredox catalysis, thus enabling C-S bond formation.[1] This knowledge, combined with the fact that post-translational modification of amino acids in peptides and proteins are of pivotal importance in chemical biology, encouraged us to attempt the visible light-induced modification of cysteine residues. Firstly, we investigated the reactivity of cysteine towards trifluoromethyl radicals.[2] After few positive results, it became clear that the idea to use cysteine to trap other radicals was worth investigating. Starting from highly electrophilic diazonium salts, we hypothesized it would be possible to generate an aryl radical through oxidative quenching of the excited state of a photocatalyst, such as Ru(bpy)32+*. To our delight, upon exposure to visible light of a mixture of diazonium salt, Ru photocatalyst and cysteine, a significant amount of the desired arylated cysteine derivative was observed.
All hands on deck
By summer 2015, our promising arylation strategy started to take shape. However, around the same period, my colleague Dario and I started to fully dedicate our time to the preparation of a review article.[3] Luckily for me, an enthusiastic and smart master student, Maarten Rubens, joined our lab and committed to the challenge of optimizing our methodology. We wondered whether the in-situ formation of diazonium salts would be compatible with our conditions. By employing t-BuONO as nitrite source and catalytic amounts of p-toluenesulfonic acid, the in situ formation of the diazonium salt, its conversion into an aryl radical, and its trapping by the thiol moiety of cysteine, were achieved in a one-pot system. Avoiding the preparation and isolation of diazonium salts proved convenient both from a time-saving perspective and, most importantly, from a safety point of view. Moreover, with Maarten’s help, we demonstrated the possibility to employ an organic dye, Eosin Y, as the photocatalyst for our transformation, therefore rendering our arylation strategy metal-free.
With the optimized conditions in hand, we started to explore the scope of different diazonium salts as coupling partners. We observed a very broad tolerance of functional groups: almost all anilines in our DUPA closet reacted in moderate to excellent yields to give the corresponding arylated cysteine derivatives! (Figure 2).

After some weeks of batch experiments and with a good scope of isolated compounds, we started to feel the urge to… go with the flow! After all, the Noёl research group is a flow group and we were curious to see what advantages microflow technology would bring to this methodology.
Maarten quickly assembled a capillary microreactor (Figure 3) consisting of PFA tubing and coiled around a homemade 3D-printed holder (if you’re interested in “microcapillary reactors for dummies” ☺ please see DOI:10.1038/nprot.2015.113 ). The efficient irradiation of the reaction mixture and the optimal mixing achievable in a microreactor resulted in a remarkable acceleration of the reaction kinetics and in increased yields. Therefore, within few weeks of hard work, we could expand our scope and report several examples of substrates showing an increased yield in flow.

Starting materials wanted!
The batch and flow results made us undoubtedly happy, but we knew that to strengthen our methodology it was time to start working on more complex model substrates. Being mainly a flow group, we didn’t have access to a nice library of frozen peptides, nor to a peptide synthesizer, so we started looking into the fastest way to obtain relevant cysteine containing dipeptides to test the applicability and the selectivity of our methodology towards other amino acidic residues. In order to reduce the synthetic steps to the minimum, while avoiding protection and deprotection steps of the amino acids side chains, we decided to prepare our dipeptides through native chemical ligation (NCL).[4]
NCL is a powerful strategy used by chemical biologists to construct large peptides or small proteins starting from two or more unprotected peptide fragments. In our case, NCL represented a straightforward methodology to easily access in two steps a small library of relevant peptides.[5] NCL proceeds through the formation of a thioester intermediate (see Figure 4: step 1) and its rearrangement (through the so-called S,N-acyl shift) to yield a native amide bond (see Figure 4: step 2).
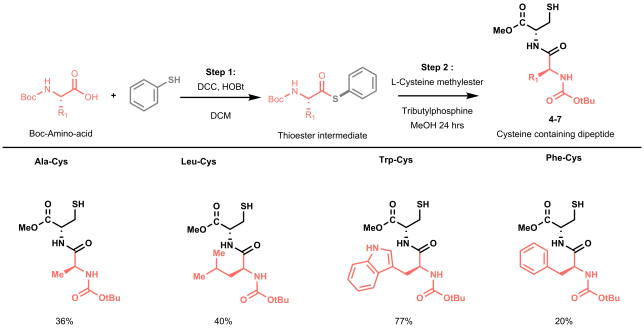
All prepared dipeptides were subjected to our batch methodology and successfully converted to the desired arylated derivative in moderate to good yields. In our eyes, these looked like satisfying results: not only did we prove the applicability of the methodology to simple model peptides, but we also demonstrated the selectivity of our protocol in presence of other amino acids that could interfere (i.e. C-2 or C-3 arylation of the indole in tryptophan).
The next level.
One of the pieces of advice that Tim gives to us PhD students is to be always critical to our own work and to put ourselves in the reviewers’ shoes. This advice sure led me to the conclusion that, despite all the nice results obtained so far, our methodology still wasn’t ready for publication. We needed more convincing results on more complex substrates, we simply needed more.
It was also clear to us that our resources and expertise in the field of peptide modifications were limited. Luckily, we knew on which door to knock to ask for some help! Annemieke Madder’s group at Ghent University had just the expertise we needed. Both Tim and I were familiar with Annemieke’s group (Tim obtained his Ph.D. at Ghent University and I did my master thesis project in the very same group!) and were enthusiast about the opportunity to collaborate with them. After a first meeting, Annemieke and one of her Postdocs, Smita Gunnoo, decided to give us a hand and to kindly host me for a short period in their lab to perform some experiments.
Smita invested some time to get to know the project and discussed with me the aspects that needed improvements in order to facilitate the application of our methodology to peptides. She prepared for me a small library of interesting peptides and together we started to look for the optimal reaction conditions. After screening several solvent systems, reaction times and sequences we finally obtained good results on the modification of a hexapeptide containing, among others, L-serine and L-lysine residues. This model peptide allowed us to demonstrate the compatibility of our cysteine-modification strategy with other commonly used post-translational modifications involving these same residues. The simplest reaction conditions turned out to be the best ones as well: a PBS buffered solution as solvent, pre-made diazonium salts and Eosin Y were sufficient to fully convert the model peptide to its arylated derivative within 30 minutes of irradiation time. With these results in hand, we finally felt confident that the project was ready for publication.
A final word.
Writing this short blog gave me the opportunity to re-think about the story of this project. As with every challenging project, the achievement of positive results was only possible after many other negative ones. This sure was a demanding work to complete, and the visible light-mediated arylation of cysteine wouldn’t have been possible without the help of all other people involved.
My gratitude goes to Maarten for his great help with the flow scope, to Smita for showing me how to make this work on peptides and to Tim for always believing in the project (and in me).
This work was funded by the Marie Curie ITN grant “Photo4Future” (check out our website: http://www.photo4future.com)
In our group, we believe in the importance of open access scientific publications, which is why this manuscript is available free of charge for all those interested!
I hope you will enjoy the reading,
Ciao!
Cecilia Bottecchia
The paper discussed in this blog was published as: Visible Light-Mediated Arylation of Cysteine in Batch and Flow. C. Bottecchia, M. Rubens, S. B. Gunnoo, V. Hessel, A. Madder and T. Noël, Angew. Chem. Int. Ed. DOI: 10.1002/anie.201706700.
References :
[1] (a) X. Wang, G. D. Cuny, T. Noël, Angew. Chem., Int. Ed. 2013, 52, 7860-7864; (b) N. J. W. Straathof, B. J. P. Tegelbeckers, V. Hessel, X. Wang, T. Noël, Chem. Sci. 2014, 5, 4768-4773.
[2] C. Bottecchia, X. J. Wei, K. P. Kuijpers, V. Hessel, T. Noel, J Org Chem 2016, 81, 7301-7307.
[3] D. Cambié, C. Bottecchia, N. J. W. Straathof, V. Hessel, T. Noël, Chem. Rev. 2016, 116, 10276-10341.
[4] E. C. B. Johnson, S. B. H. Kent, J. Am. Chem. Soc. 2006, 128, 6640-6646.
[5] L. Markey, S. Giordani, E. M. Scanlan, J. Org. Chem. 2013, 78, 4270-4277.





 A new book on “Organometallic Flow Chemistry” with Tim as editor has been published in the series “Topics in Organometallic Chemistry” and is available, in hardcover and eBook, on
A new book on “Organometallic Flow Chemistry” with Tim as editor has been published in the series “Topics in Organometallic Chemistry” and is available, in hardcover and eBook, on 


